Selbstbau - Spektrographen sind relativ preiswerte Instrumente zur schnellen Simultan - Aufnahme des gesamten Bereichs eines Probenspektrums und auch zur Licht - Sammlung über längere Zeit bei sehr intensitätsarmen Licht - Quellen. Ohne spezielle Software zur Umsetzung der Spektren - Bilder in Spektrogramme ist die Gewinnung Letzterer allerdings sehr zeitaufwändig.
Für die Vorprüfung im vorliegenden Fall wurden lediglich Rohspektren aufgenommen, von denen weder die Werte des Untergrundes bei Dunkelheit noch die einer Vergleichslösung ohne die Inhaltsstoffe der Wasser - Probe subtrahiert wurden wegen der erwähnten Umständlichkeit der hier angewandten Methode der manuellen Umsetzung in Spektrogramme.
Technik der Spektren - Gewinnung
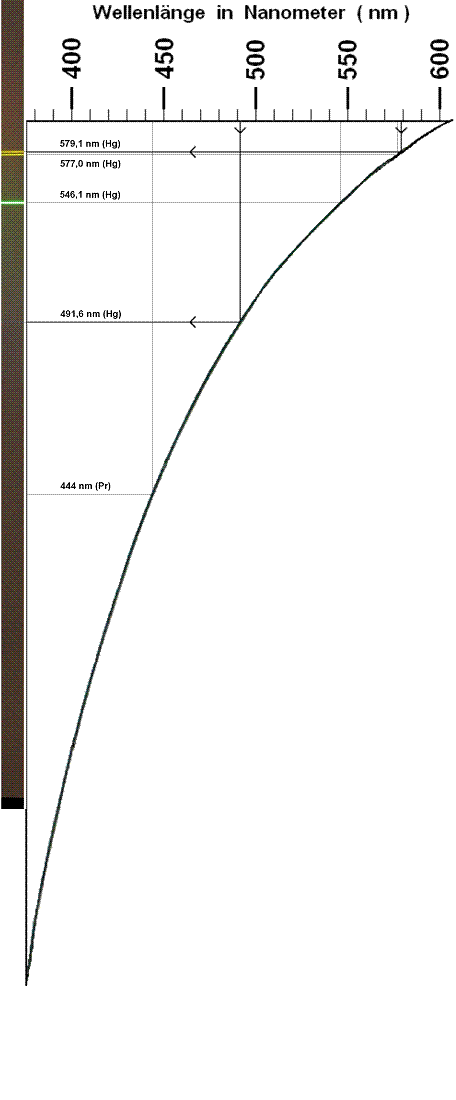
Als Instrument diente der an anderer Stelle [5] beschriebene "Selbstbau - Spektrograph 3", Prismen - Version. Der Spalt des Spektrographen wurde möglichst schmal und die verwendete Digital - Kamera "Canon EOS 450D" auf 55 mm Kleinbild - Äquivalent - Brennweite, Blende 16, 20 s Belichtungszeit und die Empfindlichkeitsstufe ISO 400 eingestellt. Der nach Stehenlassen der Analysenprobe erhaltene, farblos und klar durchsichtig erscheinende Überstand über dem Niederschlag wurde in einer Flasche aus klar durchsichtigem, farblosem Kunststoff in einer Schicht - Dicke von 6 cm vom Licht einer 100 W - Klarglas - Glühlampe durchstrahlt. Zur Wellenlängen - Kalibrierung wurde das Emissionsspektrum einer an anderer Stelle beschriebenen, mit einer Niederdruck - Quecksilber (Hg) - Dampf - Gasentladungsröhre ausgestatteten Handlampe [5][6] in das Probenspektrum eingeblendet. Zur Ermittlung der zu einer Stelle im Spektrum gehörigen Licht - Wellenlänge wurde ein Kalibriergraph - Diagramm benutzt [11], das in der für die hier untersuchte Probe bearbeiteten Form zusammen mit dem Spektrum unten abgebildet ist. Für den hiesigen Zweck wurde die Größe des Diagramms an die des Spektrums angepasst, um dessen Informationsgehalt unverändert zu erhalten. Das Diagramm ist hier gegenüber dem zur Kalibrierung verwendeten Original verkleinert und um 90 ° gedreht abgebildet, damit nicht eine Überbreite der Seite zum Horizontalscrollen zwingt.
Der Spektren - Untergrund
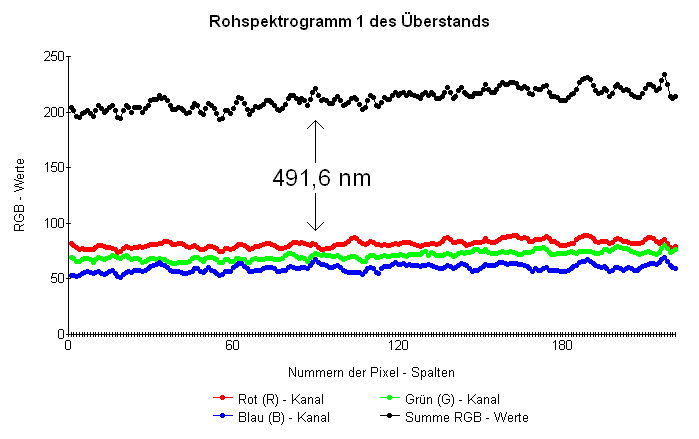
Eine Licht registrierende Vorrichtung erzeugt auch ohne Belichtung mit einer Spektrum - Quelle Signale (Untergrund - Signale), bedingt durch aus der Umgebung eingestreutes Licht (Störlicht) und, bei elektronischen Sensoren, elektrische Ströme, die auch ohne Licht - Einwirkung in den Schaltkreisen fließen. Bei aus einer Vielzahl von Einzelsensoren bestehenden elektronischen Bild - Aufnehmern bewirken herstellungsbedingt etwas unterschiedliche Eigenschaften der Sensoren und ihrer zugehörigen Beschaltungen in von ihnen gelieferten Bildern ortsabhängige Helligkeitsunterschiede (Bild - Rauschen) auch bei vollkommen gleichmäßiger Beleuchtung der gesamten Fläche des Bild - Aufnehmers. Lichtschwache Muster eines Spektrums sind auf einem ähnlich intensiven Rauschuntergrund schwer zu erkennen. Um diesen im vorliegenden Fall abzuschätzen wurden in einem Teilbereich des in "Paint" [8] geöffneten, auf senkrechte Stellung der Kalibrierspektrallinien ausgerichteten Spektrum - Photos auf einer Pixel - Zeile deren Pixel mit der "Farblupe" [7] manuell abgetastet. Zu jedem ausgelesenen Pixel wurden die Werte für die Pixel - Spalt - Nummer und die Helligkeiten im Rot -, Grün - und Blau - Farbkanal im betreffenden Pixel ermittelt und in das Tabellenkalkulationsprogramm der Software "Works" eingetragen, das aus den Daten folgendes "Rohspektrogramm 1 des Überstands" erstellte:
Das Diagramm repräsentiert einen Ausschnitt aus dem Absorptionsspektrum des Überstands der Analysenprobe im Wellenlängen - Bereich von etwa 484 nm (links) bis etwa 503 nm (rechts). Als Wellenlänge - Marke wurde der Wert 491,6 nm für die blaugrüne Quecksilber - Linie des Kalibrierspektrums im Spektrogramm markiert. In diesem zeigt sich besagte Linie als kleiner Peak mit Helligkeitsmaximum in Pixel - Spalte 89 in den Graphen des Blau - Kanals und der RGB - Summen. Nur aus dem Spektrogramm heraus wäre der Peak angesichts seiner infolge Bild - Rauschen, siehe oben, hügeligen Umgebung wohl nicht als Spektrallinie zu erkennen. Der menschliche Sehsinn jedoch nimmt an der entsprechenden Stelle im Original des Spektrum - Photos eine schwache Linie wahr. Im verkleinerten Photo in der oben abgebildeten Graphik ist sie schon nicht mehr sichtbar.
Suche nach breiten Absorptionsbanden
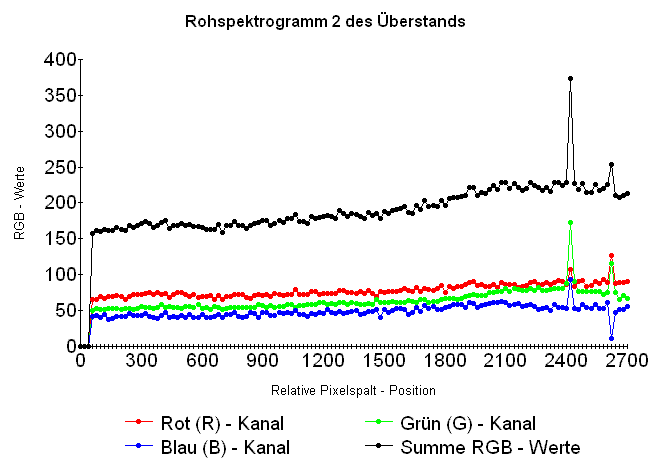
Manche Stoffe erzeugen Spektren mit breiten Absorptionsbanden, die auch in Spektrogrammen mit größeren Datenpunkt - Intervallen erkennbar bleiben. Zur Suche nach solchen Mustern wurden im Spektrum - Bild nach Öffnen im Bildbearbeitungsprogramm "Paint" [8] in stark vergrößerter Ansicht für jedes 20. Pixel einer ausgewählten Pixel - Zeile mit dem Programm "Farblupe" [7] dessen Farb- und Helligkeitsinformation ausgelesen. Zu jedem dieser Pixel wurden die Werte für die Pixel - Spalt - Nummer und die Helligkeiten im Rot -, Grün - und Blau - Farbkanal ermittelt und in das Tabellenkalkulationsprogramm der Software "Works" eingetragen, das aus den Daten folgendes "Rohspektrogramm 2 des Überstands" erstellte:
Das Übersichtsspektrogramm umfasst den Wellenlängen - Bereich zwischen etwa 400 nm (linker Rand) und etwa 600 nm (rechter Rand). Eine breite Absorptionsbande könnte hinter der schwachen Mulde in der linken Spektrum - Hälfte stecken. Schmale Banden wie die von Seltenerdelementen oder noch schmalere Spektrallinien zeigen sich nur bei zufälligem Zusammenfallen mit Abtastpixeln, wie im Spektrogramm oben nahe dem rechten Rand des Spektrogramms.
Detailspektrogramm zur Suche nach Seltenerdelementen
Da in durchschnittlichen, wie oben beschrieben mit Essigsäure behandelten Leitungswasser - Proben Stoffe fehlen, die mit Kationen der genannten Seltenerdelemente, wenn sie in Konzentrationen von wenigen Mikrogramm (µg) / L vorliegen, Niederschläge bilden, wurde hier ein Absorptionspektrum der sich beim Stehen der Analysenprobe über dem Niederschlag bildenden klaren Flüssigkeit (Überstand) aufgenommen. Inwieweit allerdings besagte Kationen trotz theoretischer Löslichkeit in den Niederschlag der Analysenprobe mitgerissen wurden, muss hier offen bleiben.
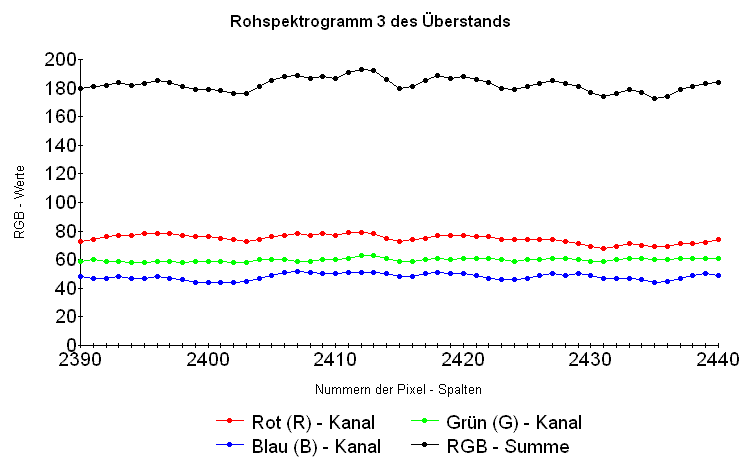
Das folgende "Rohspektrogramm 3 des Überstands", gewonnen wie bei den obigen Spektrogrammen beschrieben, zeigt im Bereich der Absorptionsbande von Pr bei 444 nm, die hier etwa bei Pixel - Spalte 2423 + / - etwa 5 Pixel liegen müsste, zwar eine schwache, aber im Rahmen des Rauschuntergrundes bleibende Senke. Die Pr - Konzentration im Überstand der Analysenprobe kann demnach höchstens einen solchen Wert haben, dass die hierdurch verursachte Schwächung des Lampenlichts in der Größenordnung der Rauschen - bedingten Schwankungen in den Spektrogramm - Kurven liegt.
Wird die von Pr ungeschwächte Licht - Intensität außerhalb des Bandenbereichs als I0 bezeichnet und die von Pr geschwächte Licht - Intensität im Zentrum der hypothetischen Pr - Bande als I, dann ist der Logarithmus zur Basis 10 des Verhältnisses I0 / I gleich dem Produkt aus dem molaren Extinktionskoeffizienten ε, der Pr - Konzentration c und der Länge d des Licht - Weges in der Probenlösung [12]:
log ( I0 / I ) = ε . c . d
Die Auflösung dieser Gleichung nach der Konzentration c ergibt folgenden Ausdruck:
c = log ( I0 / I ) / ( ε . d )
Als Wert für I0 sei hier der Helligkeitswert des Kurvenmaximums im Bereich um 444 nm, etwa 52 Einheiten, verwendet, als Wert für I das benachbarte Kurvenminimum mit etwa 47 Einheiten. In der Literatur wird für die Wellenlänge des Maximums der hier berücksichtigten Absorptionsbande des Pr bei 444 nm der Wert von ε zu 10,4 mol-1 m2 angegeben [9]. d beträgt wie oben erwähnt 0,06 m. Nach Einsetzen der Werte wird folgende Gleichung erhalten:
c = log ( 52 / 47 ) / ( 10,4 mol-1 m2 . 0,06 m )
Nach Befolgen dieser Rechenanweisung wird folgender Wert für die höchstmögliche molare Konzentration an Pr im Überstand der Analysenprobe erhalten:
c = 0,0704 mol / m3
Umrechnung in die handlicheren Maßeinheiten Mikromol (µmol), das sind 1/1000000 Mol, und Liter (L) ergibt:
c = 70,4 µmol / L
Die Umrechnung von mol in Milligramm (mg), das sind 1/1000 g, basierend auf dem Tabellenwert 140,9 g / mol [10] für die gemittelte Atom - Masse des natürlich vorkommenden Isotopen - Gemisches von Pr, führt zu folgendem Wert für c als Massenkonzentration:
c = 9,9 mg / L
Nun muss noch von der Konzentration c der Analysenprobe, die die Inhaltsstoffe der Urprobe des Leitungswassers etwa 50-fach konzentriert enthält, siehe oben, auf die Konzentration c0 in Letzterer umgerechnet werden:
c0 = 70,4 µmol / L / 50 = 1,4 µmol / L
c0 = 9,9 mg / L / 50 = 0,2 mg / L
Inhaltsstoffe in der Urprobe Stoff - Name Prüfmethode Chemisches Symbol Molare Konzentration Massenkonzentration Praseodym(III) - Kation Absorptionsspektralanalyse mit "Selbstbau - Spektrograph 3" Pr3+ Maximal etwa 1,4 µmol / L (Falls Pr vollständig im Überstand der Analysenprobe) Maximal etwa 0,2 mg / L (Falls Pr vollständig im Überstand der Analysenprobe) Auf die Aufnahme eines Spektrums im Bereich der oben erwähnten Bande von Nd bei 576 nm wurde wegen zu erwartender Überstrahlung der Bande durch die orange Hg - Linie des Kalibrierspektrums bei 577,0 nm verzichtet.